February 28, 2013
Uncovering the Mysteries of Alpha-Synuclein Natural Truncation Variants
Natalie Kukulka*
Department of Neuroscience
Lake Forest College,
Lake Forest, Illinois 60045
Download PDF
Summary
The alpha-synuclein protein is the major component of Lewy bodies, which are characteristic pathological trademarks for synucleinopathies. The aggregation of the protein has been linked to the formation of Lewy bodies and Lewy neurites in neurodegenerative diseases such as: dementia with Lewy Bodies (DLB), multiple system atrophy (MSA), Parkinson’s disease (PD); as well as formation of senile plaques in Alzheimer’s disease. Recent studies have further specified that such aggregates consist of truncated isoforms of the alpha-synuclein protein. The three alternatively spliced natural variants are: alpha-synuclein 98, 112, and 126, each of which can be classified as an in-frame deletion of a specific exon sequence in the SNCA gene. While scientists are now aware of their presence in Lewy bodies, there is still much to be discovered about the properties and roles of each truncation. Furthermore, there are many diseases linked to inappropriate alternative splicing which poses a re-consideration for the classification of synucleinopathies Thus, the purpose of this minireview is to discuss the accumulated knowledge on these truncations from the published literature up until this point.
Introduction
The primary feature recognized in the pathology of most neurodegenerative diseases is the presence of aggregated, fibrillated and insoluble deposits of a particular protein within a specific region of the brain (Beyer at al., 2005). More specifically, synucleinopathies are united by the aggregation of conformationally and posttransitionally altered alpha-synuclein, which can accumulate in various parts of the brain, such as the neocortex, hippocampus, substantia nigra, thalamus and cerebellum (Beyer, 2006). Dementia with Lewy bodies (DLB), Lewy body variant of Alzheimer disease (LBVAD), multiple system atrophy (MSA) and Parkinson’s disease (PD) can be therefore pathologically unified by the presence of intraneuronal cytoplasmic inclusions, known as Lewy bodies, with alpha-synuclein as their main component (Beyer et al., 2004, Beyer 2006, Dufty et al., 2007). Inevitably, such pathological features lead to the onset of different neurological impairments revealed through distinct clinical features (Beyer et al., 2004, Beyer 2006, Dufty et al., 2007).
The composition of Lewy bodies and the involvement of alpha-synuclein protein were vastly studied and agreed upon by many laboratories over years of experiments. Western blots revealed the presence of partially truncated and insoluble aggregates of both full-length and truncated alpha-synuclein proteins (Lucking et al., 2000, Li et al., 2005). While the nature of the truncations remained a mystery for a long time, researchers have now identified three naturally occurring truncations of the alpha-synuclein protein that is known to comprise Lewy bodies, these are: alpha-synuclein -112, -126 and -98. While the balance between these isoforms is starting to show potential for being one of the major reasons for Lewy bodies’ formation, it should be noted that overall Lewy body diseases (LB) are very heterogeneous. That means that alterations to more than just one molecular pathway may be involved in disease development. While alpha-synuclein exhibits deregulated alternative splicing there are other significant posttranslational modifications that play a role in the initiation of LB diseases (Beyer et al., 2012).
The role of this review is to familiarize you with the knowledge about the alpha-synuclein protein and present the accumulated information on its newly discovered isoforms. Moreover, I will discuss alpha-synuclein’s isoforms involvement in the pathology of neurodegenerative diseases and explore a possible link to other diseases caused by alternative splicing mishaps. While there are not many studies with animals pertaining to alpha-synuclein natural truncations I will review the latest knowledge gathered predominately from mice, as well as add valuable details from human patient studies. Lastly, I will hypothesize upon possible avenues of future research on the truncations and their predicted behavior in yeast models.
Alpha-synuclein
Synuclein protein was first isolated from Torpedo californica, or Pacific electric ray, but by now its complete sequence has been analyzed in more than 20 species with its mutations, posttranscriptional modifiers, polymorphisms and truncations (Xiong et al., 2010). While there is still a lot unknown about the alpha-synuclein protein, it appears to play a role in regulation cell differentiation, synaptic plasticity, regulation of the size of presynaptic vesicular pool, and dopaminergic neurotransmission (Luckin et al., 2000, Beyer et al., 2004). This is further supported by its history of binding to the phospholipid membranes in vitro, and co-localization with synaptic vesicles in vivo (Luckin et al., 2000). While alpha-synuclein is predominantly expressed in the central nervous system, it is not a brain-specific protein because its presence has been recognized in other tissues, such as, the heart, muscles and pancreas (Beyer et al., 2004, Beyer et al., 2006). Further research specified and confirmed its high expression in skin, lungs, kidney, spleen, heart, liver and muscle samples, with the highest expression in the brain (Beyer et al., 2008).
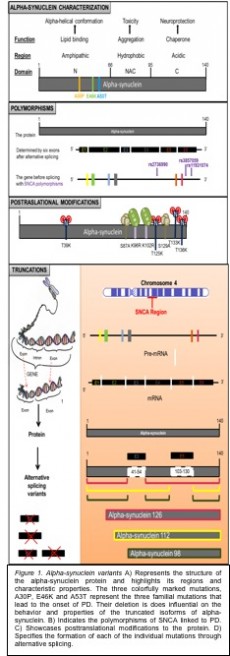
Structurally speaking, alpha-synuclein is a rather small, 140 amino acid long, acidic protein which is encoded by SNCA gene on chromosome 4q21 (Figure 1A) (Beyer et al., 2006). Its native form is to be unfolded, which increases its predisposition to self-aggregate based on varying environmental conditions making it a very dynamic molecule (Lucking et al., 2000, Beyer et al., 2006, Bisaglia et al., 2009). More specifically, in an aqueous solution it conserves its unfolded random coiled structure, but when it comes in contact with acidic phospholipid vesicles it folds into an alpha-helical structure, or forms insoluble fibrils with a high beta-sheet structure leading to formation of Lewy bodies (Beyer et al., 2004, Beyer et al., 2006). In the past, it has been showcased, through artificial truncations, that regardless of deletions of the residues 9-41, 43-56 and 58-102, the protein’s ability for binding lipids and acidic phospholipid vesicles was conserved (Lucking et al., 2000). This indicates that lipid binding must be a significant property of alpha-synuclein that is highly preserved.
Aside from environmental conditions there are molecular alterations that have severe consequences in the function of the protein. There are more than 300 different
posttranslational protein modifications some of which include: proteolysis, phosphorylation, nitrosylation, lipidation, oxidation, acylation, ubiquitination, and sumoylation (Figure 1C) (Beyer et al., 2006, Beyer et al., 2012). These molecular stimuli lead to changes in protein size, charge, structure and conformation affecting key protein characteristics (Beyer et al., 2006). Aside from the modifications, there are also natural variants of the protein that similarly alter the newly formed protein’s properties. For example, simple nucleotide alpha-synuclein gene polymorphisms can significantly affect the level of protein expression in substantia nigra and the cerebellum (Fuchs et al., 2008). Furthermore, a study published by Beyer et al. (2007) identified a new alpha-synuclein gene poly-T polymorphism, situated upstream to exon 3, which resulted in three different alleles representing various expression of a natural truncation (Figure 1B) (Beyer et al., 2007).
It is known that about 15% of alpha-synuclein structures are truncated forms, which in vitro show high tendency for aggregation (Liu et al., 2005, Beyer et al., 2012). Most specifically the known forms of truncations are due to: 20 S proteasome-mediated cleavage, cathepsin D-mediated cleavage, calpain-I-mediated cleavage, neurosin-mediated cleavage and matrix metalloproteinase 3 mediated cleavage (Beyer et al., 2012). Such methods of cleavage have resulted in truncation in both the NAC and C terminals of the alpha-synuclein protein significantly altering its behavior and leading to severe pathological consequences (Beyer et al., 2012). For example the expression of C-terminally truncated alpha-synuclein (1-120) through calpain-cleavage, lead to pathological inclusions and reduction of striatal dopamine levels in transgenic mice (Dufty et al., 2007). In 2005, it was noted that C-terminal truncated alpha-synuclein proteins induced rapid aggregation of the full length protein, while also increasing cellular vulnerability to oxidative stress (Liu et al., 2005). This was further elaborated upon in 2010, when rat studies revealed that the co-expression of C-terminal truncated alpha-synuclein (1-110) enhanced full-length alpha-synuclein induced pathology of PD (Ulusoy et al., 2010). Later on, patient studies showcased that C-terminally truncated alpha synuclein forms (1-110, and 1-119) were present in the Lewy bodies and Lewy neurites of Lewy body variant of Alzheimer disease (LBVA), and Lewy neurites in Alzheimer disease contained alpha-synuclein 119 (Lewis et al., 2010). Furthermore, in 2011, Ohrfelt and his team identified N-terminally acetylated full-length alpha synuclein, and two N-terminally acetylated C-terminally truncated forms (1-139 and 1-103) in DLB and PD patients.
Recently there has been a breakthrough in the identification of three naturally occurring truncations, due to alternative splicing in SNCA gene, resulting in alpha-synuclein-112, -126 and -98 (Figure 1D).

Alpha-synuclein 112
Alpha-synuclein 112 is an in-frame deletion that corresponds to the deletion of exon 5 resulting in a total weight of about 11.4 kDa (Beyer et al., 2006). The deletion of amino acids 103-130 results in a characteristic shortening of the C-terminal, which makes the protein highly hydrophobic and prone for aggregation. In the past it has been shown that C-terminally truncated alpha-synuclein demonstrated higher aggregation tendencies with would be consistent with alpha-synuclein 112 tendencies to form aggregates (Li et al., 2005). Furthermore, the deletion of Calpain cleavage site (117-122) by itself resulted in aggregation and of adaptation of beta-sheet configuration of the alpha-synuclein protein (Dufty et al., 2007). Since this site overlaps with the deletion in the 112 truncation, this
further explains and supports alpha-synuclein 112’s predisposition for aggregation.
Additionally, the truncation undergoes the deletion of an important phosphorylation site at serine 129 (Beyer et al., 2006, Xiong et al., 2010). The phosphorylation process has been known to enhance aggregation, but since the main phosphorylation site on the protein is absent its aggregation tendency may be possible due to increased phosphorylation on a less valuable site, serine 87, or its new structure.
As mentioned in the introduction, both genetic and environmental factors hold an influence on the function of the protein. In relation to the alpha-synuclein 112 truncation the genetic variability of the 3’ region at the SNCA gene proved to be associated with the level of susceptibility to PD and the correlation of the different genotypes with the relative levels of alternative splicing was linear (McCarthy et al., 2011). Specifically, the GG genotype, the ‘risk’ indicator, showed the highest expression levels of SNCA 112 mRNA, followed by GA, and the ‘protective’ genotype, AA, resulted in lowest expression of SNCA 112 mRNA (McCarthy et al., 2011).
In terms of environmental stimuli, in the past, it has been shown that ferric iron levels were elevated in the substantia nigra of PD patients resulting in aggregation of truncated fibrils that resembled those found in Lewy bodies (Lucking et al., 2000). When the alpha-synuclein 112 truncation was exposed to such conditions it also showed the tendency to aggregate further supporting its involvement in formation of Lewy bodies (Lucking et al., 2000).
Initially, upon the discovery of the truncation, it was believed that alpha-synuclein 112 was a brain-specific isoform (Byer et al., 2004). Nevertheless, further research identified the isoform expression at low levels in skin, lungs, kidneys, heart and muscles, but not in spleen or liver (Beyer et al., 2008). Furthermore, in terms of the brain localization, SNCA 112 levels were at low expression in the adult caudate nucleus and the fetal parietal cortex, but were highly expressed in the fetal and adult frontal cortex, adult temporal and parietal cortices and adult cerebellum (Beyer et al. 2008).
Alpha-synuclein 126
Alpha-synuclein 126 is an in-frame deletion that corresponds to the deletion of exon 3 resulting in a total weight of about 13.1 kDa (Beyer et al., 2006). The deletion of residues 41-54 results in elimination of the two familial PD mutations A30P and E36K, as well as a characteristic interruption of helices, three and four, which are responsible for protein-membrane interactions (Beyer et al., 2006, Xiong et al. 2010). In the past, it has been shown that the N-terminal sequence (1-66) had to be intact to maintain the protein’s membrane interaction properties, without which further aggregation would be hindered (Beyer et al., 2006, Bisaglia et al.. 2006). Therefore, it could be concluded that the alpha-synuclein 126 isoform has protective functions against the formation of Lewy bodies.
As previously mentioned, the poly-T polymorphism of alpha-synuclein yielded there different alleles, which were shown to correlate with the expression of alpha-synuclein 126. The shortest poly-T stretch correlated with the lowest alpha-synuclein 126 expression levels, whereas the longest correlated with the highest levels of the truncation’s expressions (Beyer et al., 2007). Additionally, the correlations proved to be age specific, with low level expression being more prominent at ages under 60, and high levels at ages over 80.
Further research also located the abundance of the alpha-synuclein 126 isomer within specific regions of the brain. The SNCA 126 levels were lower in the adult caudate nucleus and the fetal parietal cortex, but were highly expressed in the fetal and adult frontal cortex, adult temporal and parietal cortices and adult cerebellum, just like SNCA 112 (Beyer et al., 2008). Nevertheless, alpha-synuclein 126 was also present in intermediate levels in lungs, kidney, spleen, heart, and muscles, but not in skin or liver (Beyer et al., 2008).
Alpha-synuclein 98
The last identified truncation of alpha-synuclein protein is alpha-synuclein 98, which lacks both exons 3 and 5 (Beyer et al., 2008). This means that the protein lacks both familial PD mutations, A30P and E46K, and a phosphorylation site at S129 (Xiong et al., 2010). Thus, the truncation could be characterized by a 14 amino acid deletion in the N-terminal and a 28 amino acid deletion in the C-terminal, leaving the central region of the protein intact. Its further analysis showed that the truncation was the only one that was highly brain-specific and its expression varied in different areas of the brain and between fetuses and adults. More specifically, it was highly expressed in the adult temporal and parietal cortices and the fetal parietal cortex, but it was at low expression levels in adult and fetal frontal cortex, adult caudate nucleus and adult cerebellum (Beyer et al., 2008). When alpha-synuclein 98 was analyzed in different patient populations, it showed to be highly increased in pDBL and PB, moderately elevated in AD and only slightly increased in cLBD in comparison to the controls (Beyer et al., 2008). Overall, the SNCA 98 showed the lowest expression levels when compared to the other isoforms, -112, -126 and -140.
Truncations in animal models
Due to the recent nature of the discovery of the alternative splicing truncations there have not been many studies done in animal models. These isoforms are now known to be found in Lewy bodies; however the reasons behind the mistakes in their formation are still unknown. Upon my research, I was unable to find any studies in drosophila or C. elegants (some of the more common model organisms for neurodegenerative diseases) that would focus on studying alpha-synuclein 112, 126 or 98. Nevertheless, in 2011, McLean with his team demonstrated in transgenic mice overexpressing human alpha synuclein protein (ASO mice) the abnormalities with the region specific alpha-synuclein transcript levels. Upon the comparison between wild type mice (WT) and ASO mice, at two months, they observed higher neuronal levels of SNCA-140 in the ASO mice, with highest levels in cortex, striatum and ventral midbrain and lower levels in cerebellum, hippocampus and heart. When examining SNCA-126 of the ASO mice they found highest levels of the protein in the cortex, hippocampus and striatum. Overall, the expression of SNCA-126 in ASO mice was statistically greater only in the cortex, when compared to the WT mice, but there was a general trend of increased levels in all tested tissues. In terms of SNCA-112, they did not find any levels of it in the WT mice in stratum, cerebellum or the heart, while the ASO mice showed 18 fold increase of expression in the cortex, 8-fold increase of expression in the ventral midbrain and 2-fold increase of expression in the hippocampus. Alpha-synuclein 98 did not show any differences in neuronal levels of expression in WT mice, but the ASO mice showed highest expression in the cortex and striatum. When they repeated the tests at 15 months they notices that while most of the levels of expressions of SNCA-112,-126 and -98 increased in the ASO mice, the greatest increase occurred in the ventral midbrain for all truncations. While this study needs to be replicated and potentially adapted in other model organisms, it showcased the normal, adaptive and pathological propertied of alpha-synuclein splice variants.
Truncations in human patients
In 2004, as one of the earliest studies of the expression of full and truncated alpha-synuclein protein in patients, Beyer et al. showed that the total expression of alpha synuclein was significantly increased in DLB patients compared to AD patients or controls. While that simply supported the nature of the alpha-synuclein abundance in Lewy bodies, what they found interesting was that there was a twofold increase in alpha-synuclein 112 expression in DLB compared to controls, and a threefold increase in DLB compared to AD patients (Beyer et al., 2004). Furthermore, the controls, that had no neuropthological lesions, showed to have significantly higher levels of alpha-synuclein 112 in comparison to AD, and significantly overexpressed levels alpha-synuclein 140 in comparison to both, DLB and AD (Beyer et al., 2004). Therefore this study identified one of the truncated versions of alpha-synuclein that were believed to compose a large portion of Lewy bodies (Beyer et al., 2004).
In 2006, Beyer et al. shifted their focus onto the second natural truncation, alpha-synuclein 126. They found that the expression of alpha-synuclein 126 was markedly decreased in DLB, LBVAD and AD in comparison to controls, therefore showing the opposite patter from alpha-synuclein 112 (Beyer et al., 2006). Later on, Beyer et al. analyzed the SNCA 112 and SNCA 126 expression in the same frontal cortex samples with different patient populations. They observed that alpha-synuclein 126 was markedly decreased in DLB, LBVAD and AD compared to the controls, and furthermore in DLB the expression of SNCA 112 was up-regulated and SNCA 126 was down regulated. Whereas there was a fivefold decrease of alpha-synuclein 126 in DLB patients compared to controls, there was simultaneously a twofold increase in SNCA 112 . At that point it became apparent that alpha-synuclein 126 could act as the protective form, whereas alpha-synuclein 112 could be the aggregation promoting isoform. Therefore, the interruption of the protein-membrane interacting helices was partially beneficial and the shortening of the C-terminal was harmful in terms of pathology.
In comparison to alpha-synuclein 140, the 112 truncation aggregated much more easily, while 126 truncation seems to exhibit diminished aggregation propensity further indicating a possible existence of an equilibrium between the two isoforms.
Finally, in 2008, Beyer et al. analyzed the expression of the four forms of alpha-synuclein (98, 112, 126 and 140) in the frontal cortex of PD, pDLB, LBD and AD patient populations. In Parkinson disease patients they found an overexpression of all four SNCA isoform with SNCA 126 and 98 being unregulated. In Dementia with Lew Bodies pure form patients, the expression of SNCA 140 and 126 were down-regulated and SNCA 112 and 98 were up regulated, where SNCA 140, 116 and 98 were all overexpressed. In Common Lewy body diseased patients they found diminished SNCA 112 expression levels with similar pattern of SNCA 126 and 98 t those of pDLB. Lastly, in the Alzheimer patient population, they found SNCA 140 to be nearly non-existent, with SNCA 98 being overexpressed and SNCA 112 and 126 being down regulated.
In 2012, McLean et al. looked solely at PD patients and analyzed in depth the expression of SNCA -98, -112, -126 and -140 in various parts of the brain known to be affected by the disease. Looking at the frontal cortex, substantia nigra and cerebellum, SNCA 140 expression did not vary between controls, but PD patients showed an increased expression in substantia nigra. In those same regions, SNCA 126 levels were lower in the cortex of PD and controls in comparison to substantia nigra and cerebellum, and once again PD patients showed an increased expression in substantia nigra. While the expression of SNCA 112 was higher in cerebellum of PD patients than the other two regions, there was no difference between PD and control expressions within the three regions independently. The SNCA 98 expression was lower in the cortex of PD and control in comparison to the other regions. Therefore, alpha-synuclein 126 and 98 seemed to be co-localized and elevated in substantia nigra and cerebellum, while alpha-synuclein 112 expressions was elevated only in cerebellum, making the cerebellum have the highest expression of all truncations.
Link to Alternative Splicing Disorders
While Lewy Body diseases or synucleinopathies have never been classified as an alternative splicing disease there are many disorders that fall under that classification. In the past, it has been showcased that different cancers are initiated by alternative splice variants that are tumor specific (Faustino et al., 2003, Douglas et al., 2011). Furthermore, familial dysautonomia, Hutchinson-Gilford progeria syndrome, Menkes disease and myotic dystrophy are among other disorders caused by the consequences of inappropriate alternative splicing of various proteins (Faustino et al., 2003, Douglas et al., 2011). In relation to the nervous system, the onset spinal muscular atrophy, taupathies and frontotemporal lobar dementias have also been linked to incorrect alternative splicing (Tazi et al., 2008). Since the three alpha-synuclein protein truncations are also caused by mistakes in alternative splicing, there is an increased need for further research and establishment of the significance of these isoforms in synucleinopathies and LBD. It may be possible that this group of diseases could also be classified as alternative splicing diseases under the condition that they heavily rely on the presence of the three described truncations of alpha-synuclein.

Future Directions
As an undergraduate student, eager to contribute to the thus far conducted research, I would love to study the three truncations in a model that has not yet been discussed. Yeast, have the mechanisms to make, fold, and degrade proteins just like humans do, making them an ideal model to study the properties of various proteins. In order to further understand the characteristics of different truncations and the mechanism behind Lewy body formation, which is essentially just one big aggregate of alpha-synuclein truncations, we need to study the protein at a cellular level. Yeasts are thus the ideal model organisms that provide us with the necessary array of tools.
Based on the accumulated body of knowledge I would hypothesize and test specific behaviors in regards to localization, toxicity and accumulation of the protein in yeast cells. With a choice of many different yeast strains, I would most likely focus on PYES2 4741 strain of budding yeast that is known to exhibit alpha-synuclein characteristically plasma membrane binding. Additionally, I focus on PNMT1 TCP1 strain of fission yeast that shows a different pathological property, which is the formation of alpha-synuclein aggregates. The possible hypothesis in regards to the cell localization, accumulation and toxicity has been illustrated in Figure 2.
In budding yeast I hypothesize that alpha-synuclein 98 and 126 would show diffusion throughout the cell, due to the impairment of regions responsible for plasma membrane binding; while alpha-synuclein 112 would show the formation of aggregates. While the formation of such aggregates could not be visible with just a regular green fluorescent tag (GFP), we could use enhanced green fluorescent tag (eGFP) and analyze the results. For fission yeast, I would hypothesize that alpha-synuclein 112 would showcase aggregates, while alpha-synuclein 122 and 98 would show diffusion throughout the cell.
For both models, in regards to toxicity, I would hypothesize that alpha-synuclein 112 and 98 would be more toxic than alpha-synuclein 126. Based on results from patients’ data, I would expect the accumulation to be the highest for alpha-synuclein 112, followed by 126 and then 98. Nevertheless, these predictions ought to be tested and not just stated which is my very intention.
Conclusion:
Although there is a growing body of literature on the identity of the three naturally occurring alpha-synuclein truncations their specific roles in Lewy body formation are yet to be explained. While we have a good prediction of what ought to happen upon the specific deletion, the proteins arise with a structure that is not exactly comprehended. Hopefully, with further studies we will be able to further our knowledge on alpha-synuclein 112 and 126, as well as learn more about the alpha-synuclein 98 truncations. With the development of technology and medicine we should hope for future therapies for alternative splicing disorders while also battling the mysteries of neurodegeneration.
References
Beyer, K., Lao, J. I., Carrato, C., Mate, J. L., Lopez D., Ferrer, I., & Ariza A. (2004). Differential expression of alpha-synuclein isoforms in dementia with lewy bodies. Neuropathology and Applied Neurobiology, 30(6), 601-607.
Beyer, K. (2006). Review: alpaha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathoologica, 112, 237-251.
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., López, D., Ferrer, I., & Ariza, A. (2006). Low alpha-synuclein 126 mRNA levels in dementia with lewy bodies and alzheimer disease. Neuroreport, 17(12), 1327-1330.
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., López, D., Ferrer, I., & Ariza, A. (2007). A variable poly-T sequence modulates alpha-synuclein isoform expression and is associated with aging. Journal of Neuroscience Research, 85(7), 1538-1546.
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Latorre, P., López, D., Tolosa, E., Ferrer, I., & Ariza, A. (2007). A variable poly-T sequence modulates alpha-synucelin isoform expressiona dn is associated with aging. Journal of Neuroscience Research, 85, 1538-1546.
Beyer, K., Domingo-Sabat, M., Lao, J. I., Carrato, C., Ferrer, I., & Ariza, A. (2008). Identification and characterization of a new alpha-synuclein isoform and its role in lewy body diseases. Neurogenetics, 9(1), 15-23.
Beyer, K., Domingo-Sabat, M., Humbert, J., Carrato, C., Ferrer, I., & Ariza, A. (2008). Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in lewy body disease. Neurogenetic, 9(3), 163-172.
Beyer, K., & Ariza, A. (2012). Alpha-synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Molecular Neurobiology. DOI 10.1007/s12035-8330-5
Bisaglia, M., Schievano, E., Caporale, A., Peggion, E., & Mammi, S. (2006). The 11-mer repeats of human alpha-synuclein in vesicle interactions and lipid composition discrimination: a cooperative role. Biopolymers, 84(3), 310-316.
Bisaglia, M., Mammi, S., & Bubacco, L., (2009). Review: strictural insight physiological functions and pathological effects of alpha-synuclein. The FASEB Journal. 23, 329-340.
Douglas, A., G., & Wood, M., J. (2011). RNA splicing: disease and therapy. Briefings in functional genomics, 10(3), 151-164.
Dufty, B. M., Warner, L. R., Hou, S. T., Jiang, S. X., Gomez-Isla, T., Leenhouts, K. M., Oxford, J.T., Feany, M. B., Masliah, E., & Rohn, T. T. (2007). Calpain-cleavage of alpha-synuclein: connecting preteolytic processing to disease-linked aggregation. The American Journal of Pathology, 170(5), 1725-1738.
Faustino, N., A., & Cooper, T., A. (2003). Review: pre-mRNA splicing and human disease. Genes and Development, 17, 419-437.
Fuchs,J., Tichopad, A., Golub, Y., Munz, M., Schweitzer, K. J., Wolf, B., Berg, D., Mueller, J. C., & Gasser, T. (2008). Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. The FASEB Journal, 22, 1327-1334.
Lewis, K. A., Su, Y., Jou, O., Ritchie, C., Foong, C., Hynan, L. S., White, C. L., Thomas, P. J., Hatanpaa, K. J. (2010).Abnormal neurites containing C-terminally truncated alpha-synuclein are present in Alzheimer’s disease without conventional lewy body pathology. The American Journal of Pathology, 177(6), 3037-50.
Li, W., West, N., Colla, E., Pletnikova, O., Troncoso, J. C., Marsh, L., & Lee, M. K. (2005). Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proceedings Of The National Academy Of Sciences Of The United States Of America, 102(6), 2162-2167.
Liu, C. W., Giasson, B. I., Lewis, K. A., Lee, V. M., Demartino, G. N., & Thomas, P. J. (2005). A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. The Journal of Biological Chemistry, 280(24), 22670-8.
Lucking, C. B., & Brice, A. (2000). Review: alpha-synuclein and Parkinson’s disease. Cellular and Molecular Life Sciences, 57, 1894-1908.
McCarthy, J. J., Linnertz, C., Saucier, L., Burke, J. R., Hulette, C. M., Welsh-Bohmer, K. A., Chiba-Falek, O. (2011). The effect of SNCA 3’ region on the levels of SNCA-112 splicing variant. Neurogenetics, 12(1), 59-64.
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., Isacson, O. (2012). Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Molecular and Cellular Neuroscience, 49(2), 230-239.
Ohrfelt. A., Zetterberg, H., Andersson, K., Persson, R., Secic, D., Brinkmalm, G., Wallin, A., Mulugeta, E., Francis, P. T.,Vanmechelen, E., Aarsland, D., Ballard, C., Blennow, K., Westman-Brinkmalm, A. (2011). Identification of novel αsynuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochemical Research, 36(11), 2029-42.
Tazi, J., Bakkour, N., & Stamm, S. (2008). Alternative splicing and disease. Biochimica et Biophysia Acta, 1792, 14-26.
Ulusoy, A., Febbraro, F., Jensen, P., Kirk, D., & Romero-Ramos, M. (2010). Co-experssion of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. European Journal f Neuroscience, 32, 409-422.
Venda, L. L., Cragg, S. J., Buchman, V. L., & Wade-Martins, R. (2010). Review: alpha-synuclein and dopamine at the cross roads of Parkinson’s disease. Cell: Trends in Neuroscience, 33(12), 559-568.
Xiong, L., Zhao, P., Guo, Z., Zhang, J., Li, D., & Mao, C. (2010). Alpha-synuclein gene structure, evolution and protein aggregation. Neural Regeneration Research, 5(18), 1423-1428.